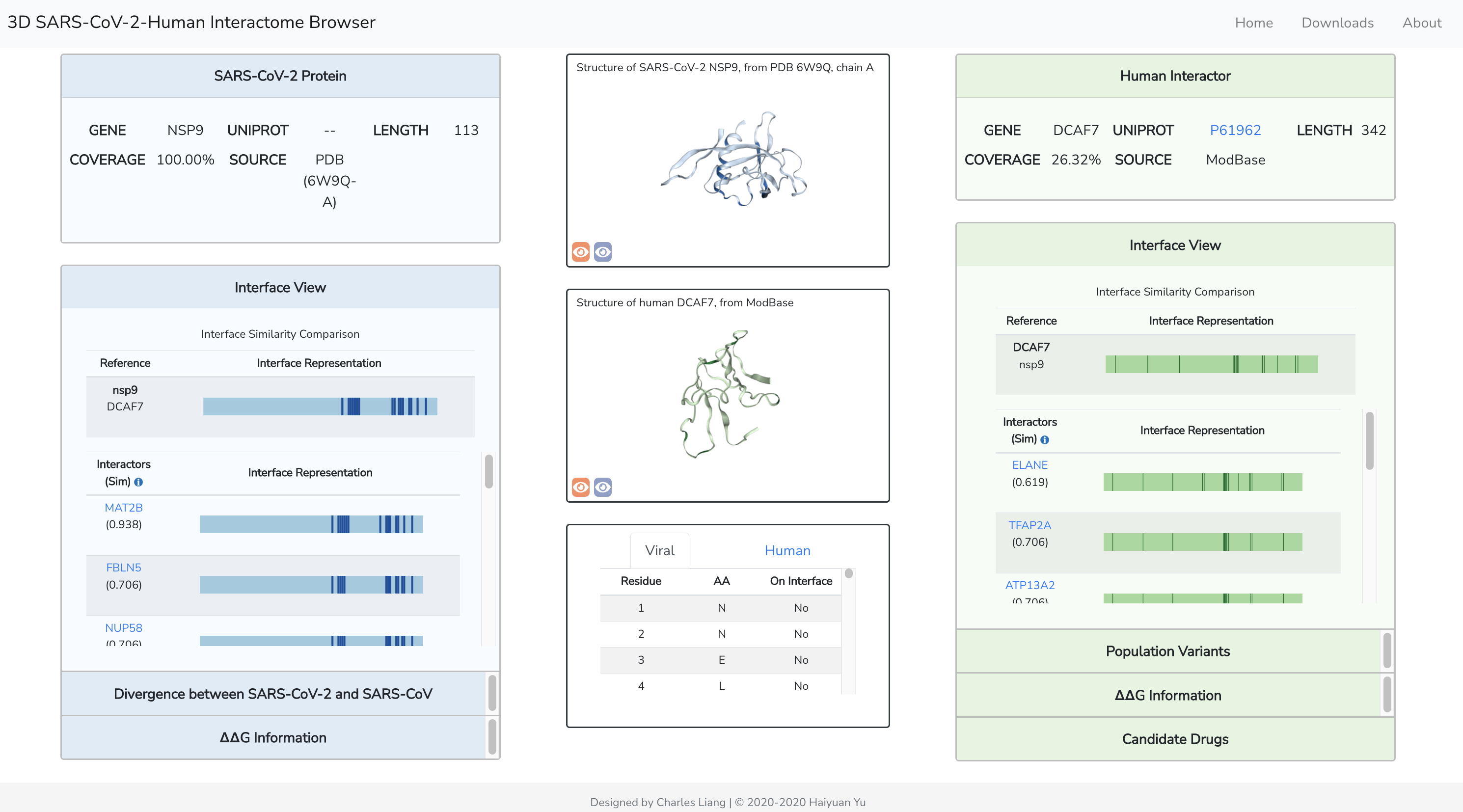
PIONEER: Protein-protein InteractiON IntErfacE pRediction
PIONEER is a deep learning-based machine learning framework for partner- specific protein interface prediction.
- Web server: https://pioneer.yulab.org
- Software and documentation: https://github.com/hyulab/PIONEER
Related paper: Xiong D et al. A structurally informed human protein–protein interactome reveals proteome-wide perturbations caused by disease mutations
Nature Biotechnology 2024.
[PDF]